Η νωτιαία μυϊκή ατροφία (SMA) είναι μια σπάνια, αυτοσωμική υπολειπόμενη νευρομυϊκή νόσος η οποία επιφέρει σοβαρή αναπηρία και χαρακτηρίζεται από την εκφύλιση των κινητικών νευρώνων και την απώλεια μυϊκής ισχύος.1,2
Κοινοποίηση σελίδας
Κοινοποίηση τώρα
ΚΛΙΝΙΚΗ ΕΠΙΣΚΟΠΗΣΗ ΤΗΣ SMA
Η ηλικία έναρξης καθορίζει το επίπεδο εκφύλισης των κινητικών νευρώνων.1,2 Η γνωστική ικανότητα δεν φαίνεται να επηρεάζεται από τη νωτιαία μυϊκή ατροφία.2
Η κλινική βιβλιογραφία δείχνει ένα ευρύ φάσμα της επίπτωσης και του επιπολασμού της νωτιαίας μυϊκής ατροφίας:
Στις Ηνωμένες Πολιτείες, η εκτιμώμενη επίπτωση της νωτιαίας μυϊκής ατροφίας είναι 8,5 έως 10,3 ανά 100.000 γεννήσεις ζώντων νεογνών.2,3
Στην Ευρώπη, η ετήσια επίπτωση ποικίλλει σημαντικά ανά χώρα και τύπο.4
Η παγκόσμια ετήσια επίπτωση ανά 100.000 γεννήσεις ζώντων βρεφών κυμαίνεται από 3,5 έως 7,1 για τον τύπο Ι, από 1,0 έως 5,3 για τον τύπο ΙΙ και από 1,5 έως 4,6 για τον τύπο ΙΙΙ.4
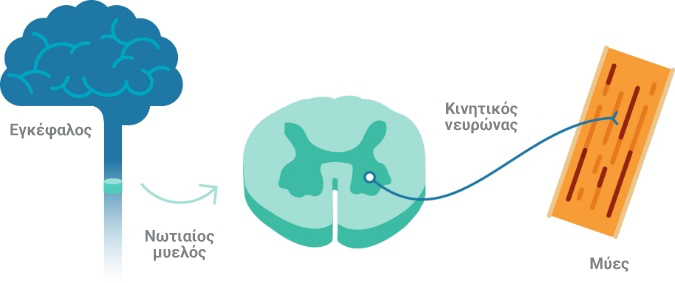
Οι κατώτεροι κινητικοί νευρώνες, που βρίσκονται στον νωτιαίο μυελό, είναι σημαντικά κύτταρα που εμπλέκονται στην κινητική λειτουργία του κεντρικού νευρικού συστήματος (ΚΝΣ)5
Μόνο για σκοπούς απεικόνισης.
Κοινοποίηση τώρα
ΓΕΝΕΤΙΚΗ
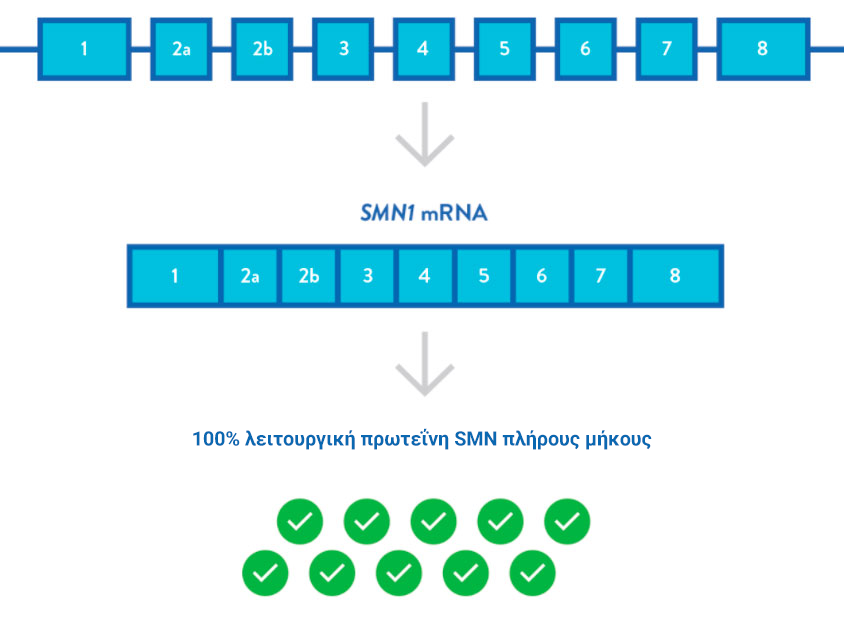
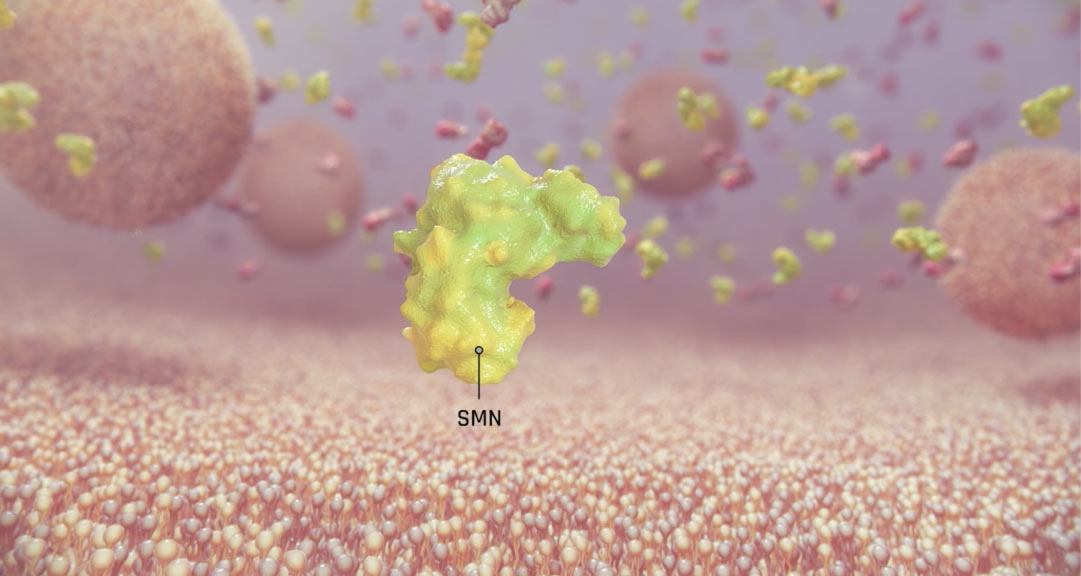
Το γενετικό ελάττωμα της υποκείμενης νωτιαίας μυϊκής ατροφίας είναι καλά χαρακτηρισμένο. Το γονίδιο επιβίωσης του κινητικού νευρώνα επιβίωσης 1 (SMN1) παράγει πρωτεΐνη SMN, που εκφράζεται στον νωτιαίο μυελό και είναι γνωστό ότι είναι απαραίτητη για την επιβίωση των κινητικών νευρώνων.2,3
Στην SMA, οι ομόζυγες μεταλλάξεις ή ελλείψεις του SMN1 γονιδίου οδηγούν σε ανεπάρκεια της SMN πρωτείνης, γεγονός που προκαλεί εκφύλιση των κινητικών νευρώνων στον νωτιαίο μυελό.7,8
Γονίδιο SMN16,7,9
Το γονίδιο SMN1 βρίσκεται στο χρωμόσωμα 5q13
Κοινοποίηση τώρα
ΓΟΝΙΔΙΟ ΕΠΙΒΙΩΣΗΣ ΚΙΝΗΤΙΚΟΥ ΝΕΥΡΩΝΑ 2
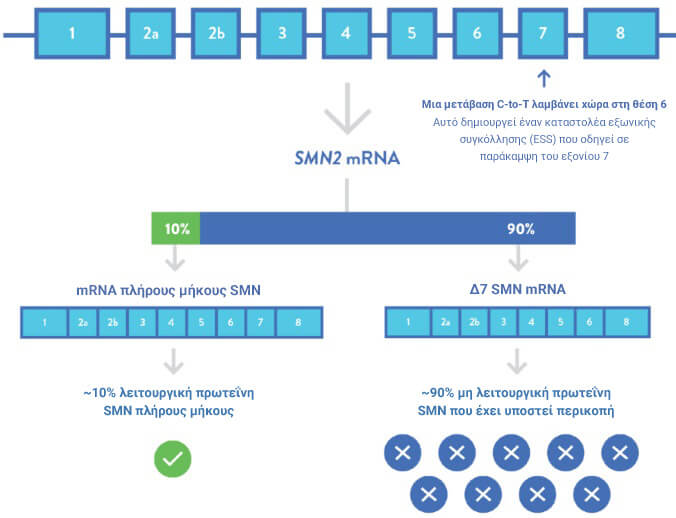
Σχεδόν όλοι οι άνθρωποι, συμπεριλαμβανομένων εκείνων με SMA, έχουν ένα δεύτερο, σχεδόν παρόμοιο γονίδιο με το SMN1, γνωστό ως γονίδιο επιβίωσης κινητικού νευρώνα 2 (SMN2).2,10
Το SMN2 είναι σχεδόν πανομοιότυπο στη γονιδιωματική αλληλουχία με το SMN1, με μόνο πέντε διαφορές νουκλεοτιδίων7
Η αντικατάσταση κυτοσίνης από θυμίνη στη θέση 6 του SMN2 δημιουργεί ένα εξωνικό καταστολέα ματίσματος (ESS) και οδηγεί στην παράκαμψη του εξονίου 7 κατά τη διάρκεια της μεταγραφής2
Αυτό έχει ως αποτέλεσμα το SMN2 να παράγει μια μικρότερου μήκους, μη λειτουργική SMN πρωτεΐνη ταχεία αποδόμησης2
Γονίδιο SMN2 2,9
Κοινοποίηση τώρα
Περίπου το 10% των μεταγραφών SMN2 οδηγούν σε πρωτεΐνη SMN πλήρους μήκους, παρέχοντας στους ασθενείς μια ανεπαρκή ποσότητα πρωτεΐνης SMN για τη διατήρηση της επιβίωσης των κινητικών νευρώνων του νωτιαίου μυελού στο ΚΝΣ.2
ΠΡΩΤΕΪΝΗ ΕΠΙΒΙΩΣΗΣ ΤΟΥ ΚΙΝΗΤΙΚΟΥ ΝΕΥΡΩΝΑ
Λόγω ανεπάρκειας του γονιδίου SMN1...1,11
Δύο αντίγραφα
Και τα δύο αντίγραφα έχουν διαγραφεί ή έχουν βλάβη
Μερική αντιστάθμιση από το γονίδιο SMN2…1,11
Ο αριθμός των αντιγράφων ποικίλλει
Μόνο το 10% της SMN είναι λειτουργικό
Κοινοποίηση τώρα
Ο ΑΡΙΘΜΟΣ ΤΩΝ ΑΝΤΙΓΡΑΦΩΝ ΤΩΝ ΓΟΝΙΔΙΩΝ SMN2 ΕΙΝΑΙ ΑΝΤΙΣΤΡΟΦΩΣ ΑΝΑΛΟΓΟΣ ΜΕ ΤΗ ΒΑΡΥΤΗΤΑ ΤΗΣ SMA
Ο αριθμός αντιγράφων του SMN2 ποικίλλει σε ασθενείς με νωτιαία μυϊκή ατροφία και μεγαλύτεροι αριθμοί αντιγράφων του SMN2 συσχετίζονται με λιγότερο βαριά νόσο:2
1 ΑΝΤΙΓΡΑΦΟ
ΠΕΡΙΣΣΟΤΕΡΟΙ ΑΠΟ ΤΟ
95%
των ατόμων με νωτιαία μυϊκή ατροφία διατηρούν τουλάχιστον 1 αντίγραφο του SMN2
Ο αριθμός αντιγράφων του SMN2 σχετίζεται με τον φαινότυπο της νόσου.
1 ή 2 ΑΝΤΙΓΡΑΦΑ
ΠΕΡΙΠΟΥ ΤΟ
80%
των ατόμων με νωτιαία μυϊκή ατροφία Τύπου Ι έχει από 1 ή 2 αντίγραφα του SMN2
3 ΑΝΤΙΓΡΑΦΑ
ΠΕΡΙΠΟΥ ΤΟ
82%
των ατόμων με νωτιαία μυϊκή ατροφία Τύπου ΙΙ έχει 3 αντίγραφα του SMN2
Όσο μεγαλύτερος είναι ο αριθμός του γονιδίου SMN2, τόσο πιο ήπιος είναι ο τύπος της νόσου.
3 ή 4 ΑΝΤΙΓΡΑΦΑ
ΠΕΡΙΠΟΥ ΤΟ
96%
των ατόμων με νωτιαία μυϊκή ατροφία Τύπου ΙΙΙ έχει 3 αντίγραφα του SMN2
Ο αριθμός αντιγράφων του SMN2 σχετίζεται, αλλά δεν είναι προγνωστικός για τη βαρύτητα της νόσου και οι αποφάσεις περίθαλψης δεν θα πρέπει να λαμβάνονται με βάση μόνο τον αριθμό αντιγράφων12,13
Σε κάθε περίπτωση νωτιαίας μυϊκής ατροφίας, ο αριθμός αντιγράφων SMN2 έχει μικρότερη προγνωστική αξία σε σχέση με την ηλικία έναρξης και τις λειτουργικές ικανότητες14,15
Εκτός από το SMN2, υπάρχουν ορισμένες ενδείξεις και άλλων γενετικών τροποποιητών που επηρεάζουν τη βαρύτητα της νόσου, συμπεριλαμβανομένων των επιπέδων της πρωτεΐνης Plastin-313
Κοινοποίηση τώρα
Λόγω του ενδεχόμενου ρόλου του στη ρύθμιση της βαρύτητας της νόσου, το SMN2 αποτελεί στόχο για διάφορες θεραπευτικές παρεμβάσεις.16
Οι χαρακτήρες που εμφανίζονται είναι πραγματικοί ασθενείς και έχει ληφθεί η απαιτούμενη συγκατάθεση για τη χρήση των ιστοριών τους από τους ίδιους και τις οικογένειές τους. Οι φωτογραφίες προορίζονται μόνο για επεξηγηματικούς σκοπούς.
Βιβλιογραφικές αναφορές
1. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet 2008;371(9630):2120-2133.
2. Darras BT, Royden Jones H Jr, Ryan MM, De Vivo DC, eds. Neuromuscular Disorders of Infancy, Childhood, and Adolescence: A Clinician’s Approach. 2nd ed. London, UK: Elsevier; 2015.
3. Lally C, et al. Indirect estimation of the prevalence of spinal muscular atrophy Type I, II, and III in the United States. Orphanet Journal of Rare Diseases 2007; 12(1): 175.
4. Jones C. PP09.1 – 2352: Systematic review of incidence and prevalence of spinal muscular atrophy (SMA). European Journal of Paediatric Neurology 2015;19(Supp 1): S64–S65.
5. Islander G. Anesthesia and spinal muscular atrophy. Paediatr Anaesth 2013;23(9):804-816.
7. Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995;80(1):155-165.
8. Medline Plus. Genetics Home Reference. SMN1. [online] 2016 Apr 20 [cited 2020 Sept 30]. Available from: URL: https://ghr.nlm.nih.gov/gene/SMN1.
9. Ogino S, Wilson RB. Spinal muscular atrophy: molecular genetics and diagnostics. Expert Rev Mol Diagn 2004;4(1):15-29.
10. Swoboda KJ. Romancing the spliceosome to fight spinal muscular atrophy. N Engl J Med 2014;371(18):1752-1754.
11. Rossoll W, Bassell GJ. Spinal Muscular Atrophy and a Model for Survival of Motor Neuron Protein Function in Axonal Ribonucleoprotein Complexes. Results Probl Cell Differ 2009;48:289-326.
13. Butchbach ME. Copy number variations in the survival motor neuron genes: implications for spinal muscular atrophy and other neurodegenerative diseases. Front Mol Biosci 2016;3:7.
14. Prior TW, Krainer AR, Hua Y, et al. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet 2009;85(3):408-413.
15. Burnett BG, Crawford TO, Sumner CJ. Emerging treatment options for spinal muscular atrophy. Curr Treat Options Neurol 2009;11(2):90-101.
16. Monani UR. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron 2005;48(6):885-896.